
填补AlphaFold3空白,字节跳动提出物理引导的方法让蛋白质动起来
世界是变化的,分子是运动的,从预测静态单一结构走向动态构象分布是揭示蛋白质等生物分子功能的重要一步。探索蛋白质的构象分布,能帮助理解蛋白质与其他分子相互作用的生物过程;识别蛋白质表面下的潜在药物位点,描绘各个亚稳态之间的过渡路径,有助于研究人员设计出具有更强特异性和效力的目标抑制剂和治疗药物。但传统的分子动力学模拟方法昂贵且耗时,难以跨越长的时间尺度,从而观察到重要的生物过程。
近年来的深度学习蛋白质结构预测模型在这个问题上也同样碰壁,往往只能预测静态单一结构,包括最近再次登上 Nature 的 AlphaFold 3,Deepmind 的研究者也承认其仍然专注于分子结构的静态预测,对动力学行为的刻画还不够。另一方面,蛋白质构象并非随机分布,而是玻尔兹曼分布,其出现的概率与其自由能量成指数级的反比。一些研究使用启发性采样或模型加噪去噪的方法,但均不能保证采样的结构是目标蛋白质的低能态,也不能保证采样的分布服从真实的玻尔兹曼分布。

造成现有模型难以预测动态构象分布的主要原因是,蛋白质结构数据集仅有实验解析的单一静态结构或结合态结构,结构数据集的偏置导致了模型难于预测真实的分布。另一方面,物理知识的缺失导致模型无法模拟分子动力学行为,从而与真实世界对齐。
在此,来自字节跳动 ByteDance Research 的研究人员提出了一种物理信息引导的蛋白质构象生成扩散模型 CONFDIFF,通过模型来预测中间时刻的力场与能量来引导模型生成高质量构象。论文已被 ICML 2024 录用。

论文链接:http://arxiv.org/abs/2403.14088
该模型充分利用了物理知识来引导模型与真实世界对齐,同时又规避了实时的力场和能量的计算,相比传统方法有巨大的加速。多项实验表明,力场和能量能够有效引导模型采样低能量的构象,进而产生更加多样化的样本,这些样本分布上也更符合真实的玻尔兹曼分布。

局部蛋白质动态

蛋白质反折叠
能量与力场引导下的蛋白质构象生成扩散模型
CONFDIFF 首先在 SE (3) 空间上构建了一个非条件的生成扩散模型,通过预测主链原子坐标和主链朝向来构建蛋白质构象。为了充分利用先验结构和序列信息,CONFDIFF 使用目标序列的预训练表示训练了一个基于序列的条件生成模型来引导上述的非条件模型,使生成构象拥有多样性的同时又能符合相应的结构与序列约束。

图 1:COFFDIFF 算法示意图
在此基础上,为使分布符合能量玻尔兹曼分布,研究者提出了能量与力场引导方法,其中最重要的是计算中间时刻的能量梯度(即力场)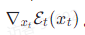 。为了规避昂贵耗时的实时能量或力场计算,研究者使用神经网络来预测这个量,并创新性提出了两种匹配训练方法并推导了相应公式,具体公式细节可参见论文。
。为了规避昂贵耗时的实时能量或力场计算,研究者使用神经网络来预测这个量,并创新性提出了两种匹配训练方法并推导了相应公式,具体公式细节可参见论文。
第一种方法是预测中间时刻能量,并使用对中间结构进行自动求导,相应的能量匹配的训练公式较为简单。但是能量可能存在数值稳定问题,而且推理结构需要储存和回传梯度,对显存和算力均存在负担。
第二种更推荐的方法是直接预测中间时刻能量的梯度(即力场),研究者也推导了相应的力场匹配训练公式。后续的系列实验也证明力场的方法更优。模型预测的能量和力场继续引导上述非条件模型,生成的构象进一步得到了的物理约束与引导,势能更低且分布更符合玻尔兹曼分布。
生成低能且服从真实分布的蛋白质构象
研究者评估了不同引导方法下模型在蛋白质构象生成任务上的性能,在快速折叠蛋白质(fast-folding proteins)和 牛胰蛋白酶抑制剂(BPTI)两种包含分子动力学模拟生成构象的蛋白质数据集上着重考察了生成样本的是否属于低能态、多样化且服从真实分布。
1. 快速折叠蛋白质(fast-folding proteins)评估
研究者在快速折叠蛋白质数据集上评估模型预测构象分布的能力。评估了生成样本和真实分子动力学样本分布之间的 Jensen-Shannon 距离 (JS Distance),以及多样性的有效性得分和 RMSF,残基之间预测接触率 RMSE。表 1 展示了各模型预测构象的性能,图 2 中展示了 TIC(time-lagged independent components)投影中的样本分布。
在预测样本分布和预测残基接触的 RMSE 等指标方面,CONFDIFF 优于已有的 EigenFold 和 Str2Str 等模型。值得注意的是,引入能量和力场引导在保持了构象多样性的同时提高了构象的生成有效性,证实了物理引导方法的优势。力场引导的方法也相对更优于能量引导的方法。

表 1:快速折叠蛋白质(fast-folding proteins)上各模型预测构象性能

图 2:快速折叠蛋白质 TIC 投影样本分布
2. 牛胰蛋白酶抑制剂(BPTI)亚稳态预测
研究者评估了模型预测 5 种 BPTI 原生折叠态附近亚稳态的质量。指标为 5 个聚类的最佳 RMSD 平均值(RMSDAVG)和最难采样的聚类 3 的 RMSD 平均值(RMSDCLS3)。如表 2 所示,CONFDIFF 在预测不同亚稳态方面有更好的能力,力场引导的模型在这两个指标上都表现最好。通过进一步比较不同采样样本量下的指标来评估模型的采样效率,如图 3 所示。CONFDIFF 模型对聚类 3 的采样效率都很好,同样地,力场引导的模型也取得了最好效果。

表 2:模型预测 BPTI 亚稳态质量
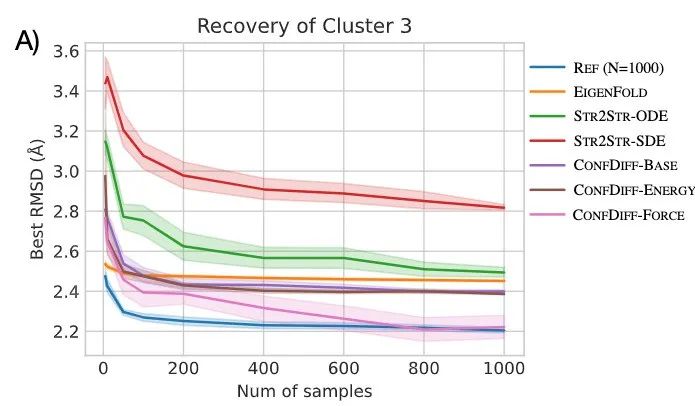
图 3:模型采样 BPTI 效率

图 4:模型预测 BPTI 亚稳态蛋白质具体例子(真实结构涂色,采样结构灰色)
3. 力场引导模型采样
研究者以快速折叠蛋白中的 WW Domain 蛋白为例,探究了 CONFDIFF 在不同程度的力场引导 (η) 和序列条件 (γ) 影响下生成构象的效果如图 5 所示。结果表明,力场引导的模型可以在不显著降低多样性的情况下提高构象稳定性。研究者同样探究了不同强度下能量引导的模型采样结果,得到了相似结论,已展示在论文附录中。

图 5:在不同的力场引导 (η) 和序列条件 (γ) 下,WW Domain 的采样构象的能量 (左) 和多样性 (右)
总结:通过物理信息引导向真实世界迈进
现有的蛋白质结构数据库的构象多样性有限,相应的蛋白质结构预测模型及在此基础上衍生的生成扩散模型都往往只能预测折叠结构而缺乏预测整个构象空间的能力。
字节跳动 ByteDance Research 的研究者首次将玻尔兹曼先验与生成扩散模型结合,使用模型预测中间时刻能量与力场并引导模型生成更加低能多样且服从真实分布的构象。这一研究有助于扩展蛋白质结构的探索从预测静态单一结构走向预测动态构象分布,迈向更真实的物理世界,为准确的药效预测、理解成药机理、设计药物、发现新靶点等提供帮助。
揭示蛋白机理,探索生命本质
ByteDance Research AI 制药团队持续在 AI for Science 方向发力
ByteDance Research AI 制药团队致力于将人工智能技术应用于科学研究与药物开发。团队在生成式蛋白质设计、蛋白质构象预测以及冷冻电镜解析等领域取得了业界瞩目的成果。
在生成式蛋白质设计方面,团队研发了基于大规模蛋白质语言模型的序列设计方法 LM-Design,大幅提高了蛋白质序列设计的准确度与效率;研发了结合扩散模型与语言模型的新一代蛋白质基础模型 DPLM,首次全面统一了蛋白质建模、理解与生成;研发了基于偏好优化的抗体设计方法 AbDPO,能够设计出同时满足多种性质和能量要求的抗体。
在蛋白质动态构象预测方面,团队研发了 ConfDiff 等模型,准确预测了蛋白质的构象变化,加深了对蛋白质生物过程的理解,还为新药研发提供了可靠的理论基础。
冷冻电镜解析方面,团队研发了 CryoSTAR 电镜解析工具,结合人工智能技术和高分辨率成像,极大地提升了生物大分子结构解析的速度和精度。这一技术有助于揭示复杂生物分子体系的构象特征和动态变化,为药物靶点的发现与设计提供了强有力的支持。
团队的研究成果多次发表在 ICML、NeurIPS、ICLR 等顶级学术会议上,得到学术界和业界的广泛认可。
© THE END
转载请联系本公众号获得授权
投稿或寻求报道:content@jiqizhixin.com
关注公众号:拾黑(shiheibook)了解更多
[广告]赞助链接:
四季很好,只要有你,文娱排行榜:https://www.yaopaiming.com/
让资讯触达的更精准有趣:https://www.0xu.cn/






 机器之心
机器之心
 关注网络尖刀微信公众号
关注网络尖刀微信公众号随时掌握互联网精彩






- 1 习近平将发表二〇二六年新年贺词 7904141
- 2 2026年国补政策来了 7808738
- 3 东部战区:开火!开火!全部命中! 7712893
- 4 2026年这些民生政策将惠及百姓 7616985
- 5 小学食堂米线过期2.5小时被罚5万 7519709
- 6 解放军喊话驱离台军 原声曝光 7428214
- 7 为博流量直播踩烈士陵墓?绝不姑息 7327605
- 8 每月最高800元!多地发放养老消费券 7238391
- 9 数字人民币升级 1月1日起将计付利息 7141831
- 10 2026年1月1日起 一批新规将施行 7040675