疾病治疗和新药研发当中,多尺度建模发挥着重要作用。而当传统科学计算方式遇到生物医药这类没有方程式可寻的问题时,亟待 AI 技术的加持。
9 月 2 日,在 2022WAIC 上海生物计算论坛上,华为中央软件院昇思MindSpore 开源项目架构师王紫东发表主题演讲《MindSpore AI + 科学计算实践》。演讲中,他主要介绍了传统生物计算领域中的发展瓶颈以及 MindSpore 在这方面所做的工作。以下为王紫东在 2022WAIC 上海生物计算论坛上的演讲内容,机器之心进行了不改变原意的编辑、整理:大家下午好!很高兴有机会和大家一起分享 MindSpore 在 AI + 生物计算方面做的实践。MindSpore 之前是传统的 AI 框架,目前正向 AI + 科学计算融合的通用计算架构演进。总体上讲,我们的实践是在高毅勤老师课题组指导下,共同合作完成的。研究中,我们主要从基于力场的分子模拟入手。借助分子模拟技术,我们可以模拟化学反应、蛋白质折叠以及同一物质在不同状态下的相变等;在行业之中,分子模拟也是比较重要的技术,如材料设计、药物设计、化工设计等场景都离不开分子模拟。同时,基于力场的分子模拟也是微观世界探索中的关键问题。针对这样的问题,研究者做了很多的探索,目标是在更大的空间尺度和时间尺度上得到精确的分子模拟,以揭示微观规律。针对微观世界,最有效的武器即为量子力学。保罗狄拉克曾说,「在有了量子力学之后,大部分物理化学现象的基本定律是完全已知的,困难在于实际应用中方程过于复杂无法求解。」量子力学本质上是求解薛定谔方程,但薛定谔方程没办法处理较复杂的体系,只要粒子数大于 2 就没有办法求解。所以只能进行近似求解。学界为了针对更大体系进行分子模拟,发展出了很多方法,如分子动力学、粗粒化动力学和连续介质力学。虽然能模拟的分子体系越来越大,但是精度在逐渐下降。以分子动力学为例,它利用第一性原理数据或者实验数据进行参数拟合,得到体系势能函数,也就是我们常说的分子力场。这种方法算得快,但精度很受限制,没有办法模拟精确的化学反应。事实上,分子模拟一直处于模拟精度和效率无法兼得的状态,我把它归结为维度灾难。在这样的瓶颈下,现在已经有一些方法在解决这个问题,最主要的就是 AI。尤其是机器学习在解维度灾难问题上很有经验,也产生了一定结果,所以传统科学计算领域以后可能会成为 AI 的主战场。现在,AI 和分子模拟已经开始进行融合,并且产生了有突破性的工作。这里我们看两个案例。第一个案例 DeePMD,它主要是用 AI 神经网络去拟合分子力场,训练数据来自于第一性原理计算数据。DeePMD 兼顾精度和性能,并且获得了 2020 年的戈登贝尔奖。案例二是 AlphaFold2,这是非常有突破性的成果,它更偏向 AI 思路,可以从海量数据中直接学习序列至结构的高维映射,效率比较高。令人惊喜的是,它的精度可以与实验精度相媲美,并被评为十大科技进步之首。AI 方法已经改变了传统计算的范式,成为一种新的可能性,软件上也要有相应的支持。我们来看一下现有软件能不能较好地支持 AI 新的范式,其实答案可能并不是很乐观。主流分子模拟软件有这么几个特点:第一是开发时间长,用户依赖性强;第二是几乎都由西方发达国家开发,由中国人开发的模拟软件极少且用户有限;第三是模拟框架老旧,灵活性差,如需添加新算法,往往需要对程序代码进行大幅改动;第四是程序多用 C/C++,甚至 Fortran 语言编写,难以兼容目前主流以 Python 为前端语言的 AI 框架。要想支持这种新范式,可能还是有很多路要走,现在做得并不是特别好。在这样的背景下,MindSpore 和高毅勤老师课题组一起开发 MindSPONGE,我们把它称之为新一代分子模拟软件。这是我们已知的第一个根植于 AI 计算框架中的分子软件,打破了传统 AI 和传统分子模拟计算的界限,并对它们的架构做了统一化的建模,取得了较好的效果。我们发现,分子模拟和深度学习的训练虽然看着好像没什么关系,但实际上有相似的计算逻辑。比如,深度学习训练是要优化一个损失函数,分子模拟可能跟它是差不多的,只不过固定的是参数,参数指的是分子力场,再去优化以空间坐标为主的样本。进行模块化拆分后,我们把 MindSPONGE 分为三大模块:系统建立、力场配置和迭代更新。我们有这么几个优势:1. 易用,无需推导原子受力的解析表达式;2. 高效,天然支持高通量模拟;3. 模块化,兼容 AI 势能函数。这里举几个分子动力学的案例。分子动力学主要是知道分子下一步怎么动,这里面的核心就是分子力场。不管是构建分子力场,还是使用分子力场,MindSPONGE 都提供了非常方便的使用接口。比如,克莱森重排有七元环与三元环转换的动力学过程,用我们自己训练的 AI 力场,以第一性原理精度去做动力学的迭代,精度及效率会优于传统方法。另外,我们对新冠病毒 Delta 变异毒株也进行了模拟。模拟结果发现,Delta 变异时,其氢键数目和接触面积相对野生型毒株有一定增加,这可能就是 Delta 毒株传播力强的原因。我们整体依托昇思MindSpore,使能 MindSpore 核心技术图算融合。MindSPONGE 的性能相较于其它框架有一定的优势。?
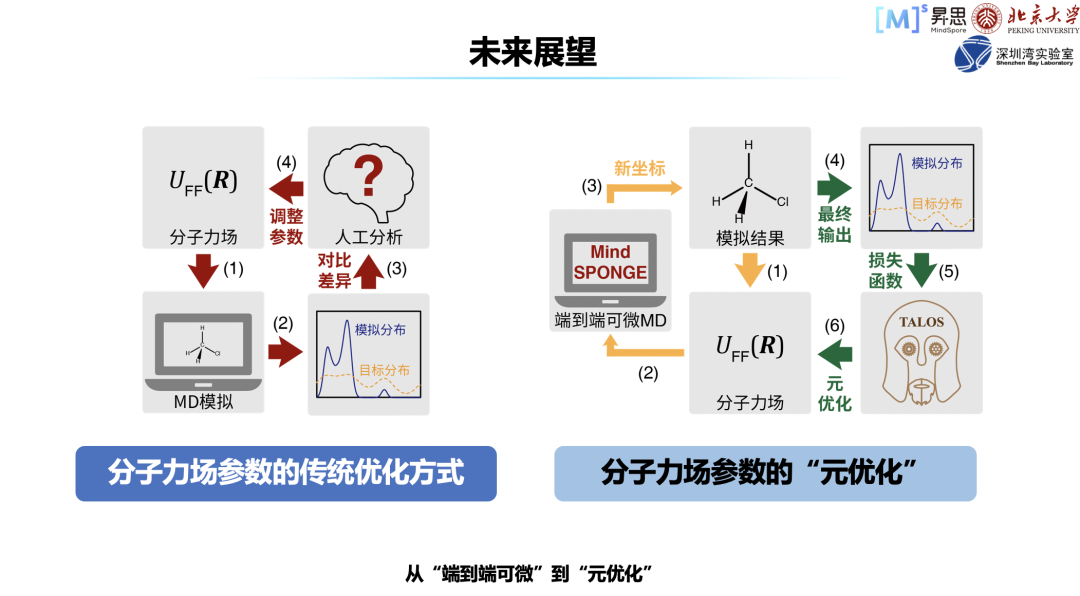
DeepMind 开源了 AlphaFold2,这对于业界在该领域发展具有巨大的推进作用,但是其并没有开源自己的训练代码,也没有提供对应的数据集。同时,对于开源出来的网络权重,其有着非商用的 License。对于科研而言没有风险,但对于商业应用的话,存在一定的侵权的问题。算法层面主要有两个问题,问题一:AlphaFold2 依赖于共进化信息,当共进化信息缺失严重时,蛋白结构预测精度会大幅下降。问题二:当存在多个模型推理出来的蛋白结构时,没有统一的算法评估这些结构的质量,这会导致无法选择最优的结构。软件层面的问题主要为算法的内存消耗,算法占用的显存与序列长度是立方关系,如果基础软件内存优化做得不好,很容易导致显存爆炸。同时,推理时性能主要消耗在共进化信息检索。针对上述不同层面的问题,我们协同高毅勤老师课题组一起进行了一些探索。?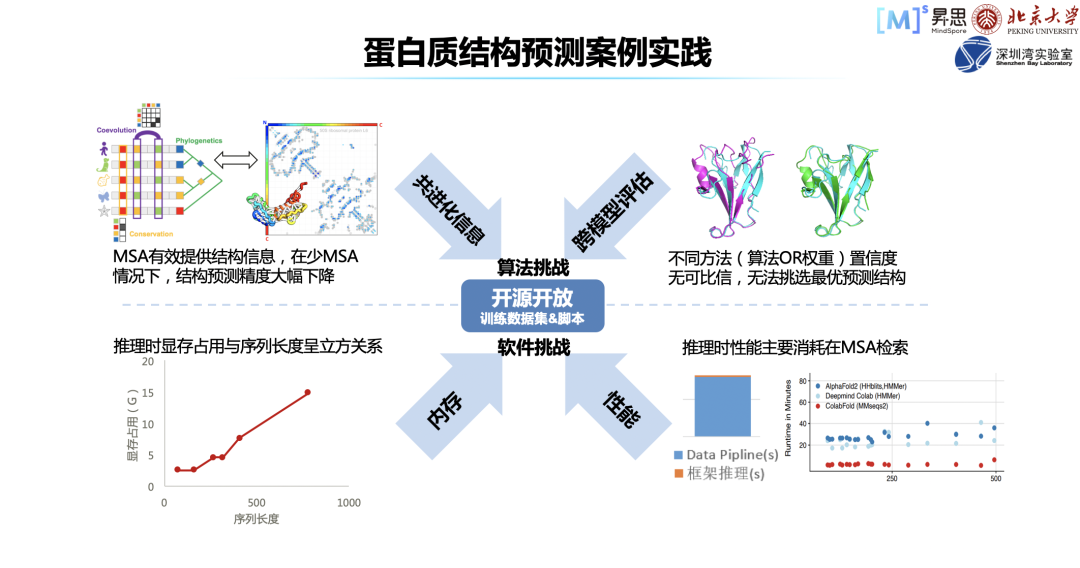 首先,我们复现了 AlphaFold2 的推理和训练,还构建了数据集,并把它开源出来,大家可以基于此数据集训练自己的模型。对于处理性能,如最耗时的前端搜索 MSA,我们都进行了优化,这项工作我们是在 4 月份完成的,当时去参加了 CAMEO 比赛,取得了较好的结果。第二步,针对算法上依赖共进化信息的问题,我们利用了人工智能领域生成模型的思路,同时借鉴了 diffusion model 的模型构建思路,针对一些孤儿序列,构建了对应的 MSA 信息,取得了较好的效果。最后,我们在蛋白质结构如何评估、如何挑出最好的蛋白质方面做了一些工作,表现也是良好的,比赛成绩优异。这项工作会在近期进行开源,如果大家感兴趣可以关注一下。蛋白质结构预测和分子动力学这两个案例有意思的地方在于,可以联动。很多时候,结构并不符合物理的规律,可能很多蛋白质是可以继续优化的,很多位置是有冲突不合理的,这时需要分子动力学模拟来做进一步的动作,也就是蛋白质结构松弛。当前 AlphaFold2, 在网络推理使用 JAX,分子动力学过程则使用 OpenMM 进行。由于 MindSPONGE 有统一建模的体系和工具,能够把蛋白质结构预测做一体化的推理,会给出更合理的结构。此外,模型的内存和性能还有很多值得优化的地方,MindSPONGE 也提供了相应的解决方案,我们称之为 SOMAS 技术。该技术主要是并行拓扑内存约束优化器,可以对所有内存进行统一统筹管理,效果还是比较明显的。性能方面,MindSPONGE 具有图算融合的特性,可以把小的细碎算子融成大算子,模型的整体性能会因此有提升。这里,对 MindSPONGE 的未来进行一些展望。基于分子力场的模拟,分子力场肯定是最重要的。传统优化方式主要还是靠人工经验,比如先有力场,就可以做环境的模拟,模拟出的分布很可能不符合实验结果,这样就会产生误差;传统方式是通过人进行分析,是不是有些参数可以改一改,再去尝试一下。相对来讲,这样比较盲目。MindSPONGE 采用的则是元优化方式。现在,我们还有很多东西没有做完,后续希望把流程都打通,也希望和大家一起合作构建。这里感谢高毅勤老师课题组,也感谢所有社区贡献的小伙伴们!最后, MindSpore 不只针对生物计算方面有相应的实践,我们在汽车、能源、气象、航空航天、EDA、材料、金融等方面都有规划。希望通过支撑更多行业应用,帮助合作伙伴商业落地,完善我们国产基础软硬件的能力。目前已经有的,主要是电磁仿真、流体仿真、分子模拟。我们并不懂行业的技术,如果没有行业专家的参与,我们根本不知道做的这些东西对不对,所以希望和大家多多合作。如果大家感兴趣,可以随时联系我,谢谢大家!
首先,我们复现了 AlphaFold2 的推理和训练,还构建了数据集,并把它开源出来,大家可以基于此数据集训练自己的模型。对于处理性能,如最耗时的前端搜索 MSA,我们都进行了优化,这项工作我们是在 4 月份完成的,当时去参加了 CAMEO 比赛,取得了较好的结果。第二步,针对算法上依赖共进化信息的问题,我们利用了人工智能领域生成模型的思路,同时借鉴了 diffusion model 的模型构建思路,针对一些孤儿序列,构建了对应的 MSA 信息,取得了较好的效果。最后,我们在蛋白质结构如何评估、如何挑出最好的蛋白质方面做了一些工作,表现也是良好的,比赛成绩优异。这项工作会在近期进行开源,如果大家感兴趣可以关注一下。蛋白质结构预测和分子动力学这两个案例有意思的地方在于,可以联动。很多时候,结构并不符合物理的规律,可能很多蛋白质是可以继续优化的,很多位置是有冲突不合理的,这时需要分子动力学模拟来做进一步的动作,也就是蛋白质结构松弛。当前 AlphaFold2, 在网络推理使用 JAX,分子动力学过程则使用 OpenMM 进行。由于 MindSPONGE 有统一建模的体系和工具,能够把蛋白质结构预测做一体化的推理,会给出更合理的结构。此外,模型的内存和性能还有很多值得优化的地方,MindSPONGE 也提供了相应的解决方案,我们称之为 SOMAS 技术。该技术主要是并行拓扑内存约束优化器,可以对所有内存进行统一统筹管理,效果还是比较明显的。性能方面,MindSPONGE 具有图算融合的特性,可以把小的细碎算子融成大算子,模型的整体性能会因此有提升。这里,对 MindSPONGE 的未来进行一些展望。基于分子力场的模拟,分子力场肯定是最重要的。传统优化方式主要还是靠人工经验,比如先有力场,就可以做环境的模拟,模拟出的分布很可能不符合实验结果,这样就会产生误差;传统方式是通过人进行分析,是不是有些参数可以改一改,再去尝试一下。相对来讲,这样比较盲目。MindSPONGE 采用的则是元优化方式。现在,我们还有很多东西没有做完,后续希望把流程都打通,也希望和大家一起合作构建。这里感谢高毅勤老师课题组,也感谢所有社区贡献的小伙伴们!最后, MindSpore 不只针对生物计算方面有相应的实践,我们在汽车、能源、气象、航空航天、EDA、材料、金融等方面都有规划。希望通过支撑更多行业应用,帮助合作伙伴商业落地,完善我们国产基础软硬件的能力。目前已经有的,主要是电磁仿真、流体仿真、分子模拟。我们并不懂行业的技术,如果没有行业专家的参与,我们根本不知道做的这些东西对不对,所以希望和大家多多合作。如果大家感兴趣,可以随时联系我,谢谢大家!
??THE END?
转载请联系本公众号获得授权
投稿或寻求报道:content@jiqizhixin.com
关注公众号:拾黑(shiheibook)了解更多
[广告]赞助链接:
四季很好,只要有你,文娱排行榜:https://www.yaopaiming.com/
让资讯触达的更精准有趣:https://www.0xu.cn/











 机器之心
机器之心
 关注网络尖刀微信公众号
关注网络尖刀微信公众号




