来源:集智俱乐部
作者:J. Pereira & T. Schwede
译者:刘培源?
审校:赵雨亭
编辑:邓一雪?
导语
以AlphaFold 2和RoseTTAfold等方法为代表的深度学习技术,不仅实现了蛋白质结构预测领域的突破性进展,也正在开启深度学习时代的相互作用组学研究。最近一篇发表在Science的工作,基于深度学习技术,创建了能够同时预测蛋白质-蛋白质相互作用的三维结构的新方法,并为作用过程建模。借助该技术,研究者甚至发现了许多此前从未观测到的相互作用。深度学习正在将以相互作用组学为代表的结构生物信息学推向新高度。研究领域:深度学习,蛋白质结构,蛋白质互作,结构生物信息学Interactomes in the era of deep learninghttps://www.science.org/doi/10.1126/science.abm8295
1. 传统蛋白互作与计算建模的加入
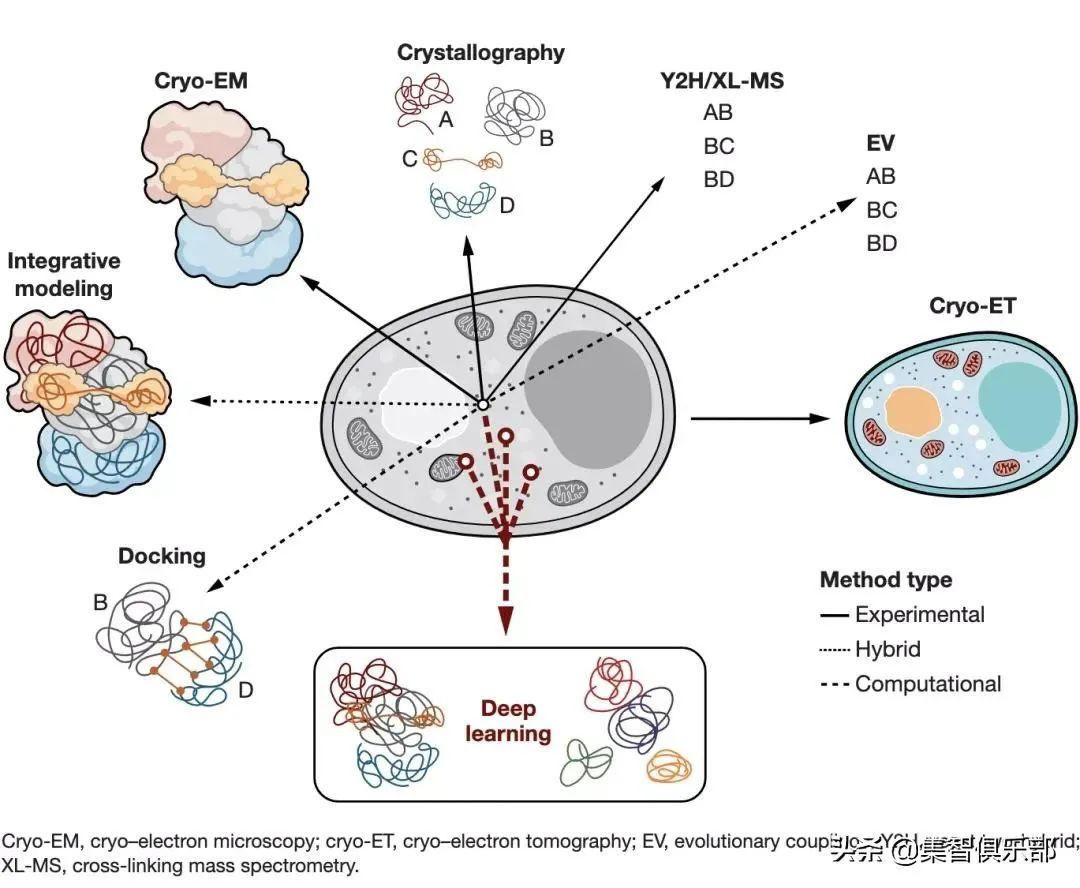
表征大分子相互作用可以更好地理解细胞内部运作。但是,如今所有可用的方法都有其局限性:一些方法告诉我们两个大分子是否相互作用,另一些方法提供关于相互作用对象的原子细节,或者仅仅是没有细胞背景的孤立组件的结构。Humphreys等人最近发表于Science论文[1]描述了一种新的计算方法,它建立在结构生物信息学中正在进行的深度学习革命[2,3]的基础上,同时预测蛋白质-蛋白质相互作用的三维结构的组成与建模。他们将其方法应用于真核系统——面包酵母菌,并预测和准确模拟了1500多种蛋白质-蛋白质相互作用,其中有106种是第一次实现。这为整个细胞的高通量、高精度建模铺平了道路。测定大分子在3D结构层面上的相互作用提供了有关大分子机制的重要信息,这些信息可以用于药物开发或生物技术开发等。高分子结晶(MX)和高分辨率低温电子显微镜(cryo-EM)等实验结构生物学方法,提供了大分子结构及其组装的原子级的细节[4]。这样的实验操作复杂:需要从细胞环境中提纯大分子。虽然诸如酵母双杂交(Y2H)和交联质谱(XL-MS)等技术允许大规模检测相互作用对象,但定点诱变或F?rster能量共振转移(FRET)实验等方法能够表征单个相互作用与界面。这些信息可以用来指导装配体建模,例如通过分子对接的综合(或混合)方法,将来自低分辨率实验的各种数据类型与计算建模相结合,以生成大分子装配体的3D表示[5]。近年来,结构生物学的视野因为结构预测计算技术而急剧扩大(如下图所示),这得益于机器学习算法的发展[6]以及开放数据库中实验信息的快速增长。例如蛋白质数据库(今年庆祝成立50周年)。自1994年以来,结构预测的临界评估(CASP)实验为检测蛋白质结构预测方法提供了一个平台,并且在其发展中经历了(也激发了)数次革命[7]。例如,用于检测远程同源关系的灵敏方法促进了基于同源性的建模,并且协同进化信息的使用进一步改进了没有已知结构同源物的蛋白质的模型。后一种方法是基于进化耦合的思想,从多序列比对中提取的耦合信号可以用来预测三维空间中的密切接触。这不仅被证明对蛋白质3D结构预测很有用,而且很容易扩展到分子间相互作用的领域:例如一种快速和准确的方法来筛选和预测蛋白质相互作用对(protein-interacting pairs,如大肠杆菌的蛋白质组)[8,9]。2. 深度学习助力预测蛋白质互作
今年,结构生物信息学有了新突破,崭新时代开启[2,3]:DeepMind的AlphaFold2算法[6]成为第一个在CASP竞赛[10]中能够对单个蛋白质结构达到接近实验原子精读的计算方法[10]。其成功基础是综合使用了最先进的深度学习方法、大规模计算能力以及过去50年积累的海量结构和序列数据。这促进了科学社区中快速而激烈的活动——RoseTTAfold作为AlphaFold2的学术竞争对手迅速崛起[11]。这两种方法都使用了最先进的深度学习方法,但是它们的核心架构不同。尽管如此,两者重要的共同点是使用了多序列比对的进化耦合(evolutionary couplings from multiple sequence alignments)——在其底层往网络中得到有效处理,从而预测原子间接触,并根据氨基酸序列准确计算目标蛋白质中原子的3D坐标。鉴于此类信号在识别蛋白质间相互作用方面已取得成功[8,9],因此探索此类方法对于在原子水平上改进蛋白质间相互作用及其组装的预测和建模是有意义的。研究大分子机器方法示意图。基于深度学习方法补充了实验技术,允许对蛋白质组装进行全蛋白质组的预测和建模虽然大多数的努力集中在改造AlphaFold2和RoseTTAFold工作流程以模拟已知组成和化学计量的蛋白质复合物[12],但Humphreys等人将RoseTTAFold的接触预测算法的速度与AlphaFold2折叠引擎的高精度相结合,并提出了一种新方法来同时准确预测和建模面包酵母菌蛋白质组中的蛋白质相互作用对——这是第一个被以如此高通量方式对其相互作用进行建模的真核生物。Humphreys等人扫描了大约800万对蛋白质,根据强烈的协同进化信号预测了哪些蛋白质之间更可能发过相互作用,并且通过连接对(joint pair)的蛋白质结构预测取代大分子连接(macromolecular docking),以模拟组装的3D结构。该方法能够准确预测1500多个相互作用对的组成并对其结构建模。这些相互作用对几乎涵盖了所有关键的真核细胞过程,包括106种未被描述的组合(可能会突显出此前未知的生物过程),以及600多个此前已知的相互作用对(根据低分辨率生物物理数据)。Humphreys等人的工作向高分辨率的整个细胞模型迈进了一步,并且已经启发了对人类线粒体相互作用的进一步研究[13]。目前,如高分子结晶和电子显微镜(EM)方法可以提供单个大分子机器的高分辨率原子表示。细胞低温电子断层成像有可能提供大分子相互作用网络的详细快照,但迄今为止只能获得亚纳米分辨率[14]。基于人工智能的高精度蛋白质组相互作用模型,可能能够及时弥补分辨率差距——尤其是对于更复杂的生物体。尽管如此,AlphaFold2和RoseTTAFold等方法提供了静态模型,未来还需要解决大分子组装的瞬态和动态特性。这项工作还突出了开放科学和基于社区的方法开发的成功。由一家商业公司开发的AlphaFold2,向这个科学社区公开模型及其源代码。这促进了不同目标基于AI的生物信息学方法的快速发展,例如Humphreys等人的研究。基于人工智能方法显然正在促进未来生命科学研究方式的转变,其中3D计算模型将经常激发新的可实验验证的假说。
未来智能实验室的主要工作包括:建立AI智能系统智商评测体系,开展世界人工智能智商评测;开展互联网(城市)大脑研究计划,构建互联网(城市)大脑技术和企业图谱,为提升企业,行业与城市的智能水平服务。每日推荐范围未来科技发展趋势的学习型文章。目前线上平台已收藏上千篇精华前沿科技文章和报告。
??如果您对实验室的研究感兴趣,欢迎加入未来智能实验室线上平台。扫描以下二维码或点击本文左下角“阅读原文”

关注公众号:拾黑(shiheibook)了解更多
[广告]赞助链接:
四季很好,只要有你,文娱排行榜:https://www.yaopaiming.com/
让资讯触达的更精准有趣:https://www.0xu.cn/


 关注网络尖刀微信公众号
关注网络尖刀微信公众号











 人工智能学家
人工智能学家