
辉瑞疫苗通过FDA认证,新冠疫情真的能被抑制吗?
12月11日,继英国之后,美国FDA宣布批准紧急授权美国辉瑞公司和德国的BioNTech公司联合开发的新冠病毒疫苗用于16岁以上的人群接种,黑暗隧道中挣扎的世人终于看到了一丝亮光。
作为FDA批准后的下一步,美国疾控中心CDC将会在明天或者后天召开免疫咨询委员会的专家讨论会,投票决定是否推荐CDC批准全面推广接种。通常CDC委员会会尊重FDA的推荐授权。所以不出意料的话,下周一就会在美国优先人群中开始进行接种了。

一位药剂师正在为辉瑞新冠疫苗针剂做标签 |?https://www.sciencemag.org
速度就是胜利
FDA的此次神速批准意味着两个历史性的突破。
从疫苗开发到批准临床使用,只花了不到一年的时间,这打破了所有历史记录。此前有不少行内专家都不认为一年之内批准新冠肺炎病毒疫苗存在可能性。要知道,以前的疫苗从开发到批准都花了数年时间,甚至数十年。
举几个例子,天花病毒从开发出第一个疫苗到普遍接种,花了154年时间。普通流感病毒疫苗从开发到使用花了15年。脊髓灰质炎疫苗用了20年。水痘疫苗用了25年。乙肝疫苗用了16年。人类乳头瘤病毒疫苗用了26年。而新冠疫苗的一切流程都被缩短到了一年之内,从认识到前人未知的新的病毒,分离测序,设计,合成,前期试验,临床三期试验,提请申请,到批准临床使用,不到12个月,这是前所未有的突破。这一切都得益于生物学技术在这个世纪的突飞猛进。
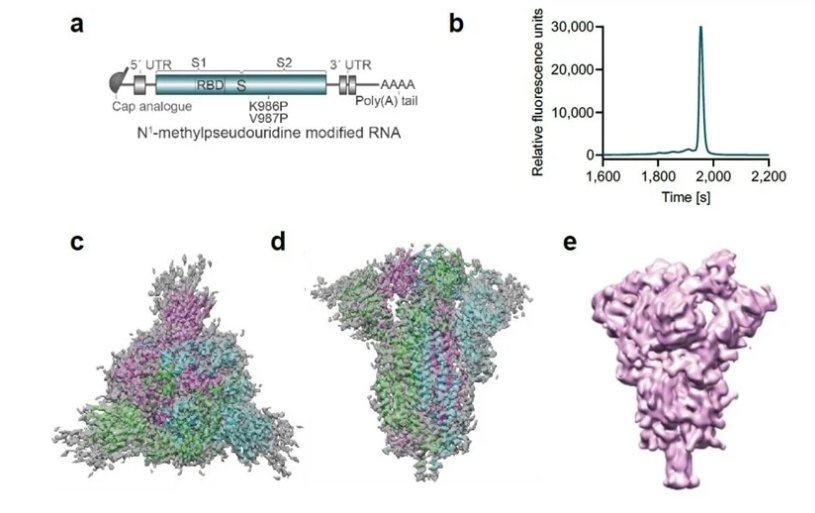
辉瑞疫苗中的抗原结构 |?https://www.sciencemag.org
这是第一个mRNA疫苗。疫苗有很多种。用病毒本身做的疫苗有减活疫苗和灭活疫苗。还有用病毒的自身的片段,和部分蛋白成分合成的疫苗。近些年有一些用病毒的DNA序列合成的腺病毒载体疫苗。但还没有一个mRNA疫苗被批准临床使用过。
此前,在moderna和辉瑞宣称在开发mRNA疫苗的时候,很多业内人士都不看好,认为成功的机会非常的小。所以此次该种疫苗获批,不但出乎许多人的意料,更是会为同类疫苗打开大门。正在观望的其他科研小组也会得到百倍的信心。
毕竟,mRNA疫苗能够成功,它的诸多优点,比如开发时间短、合成只需要病毒的基因序列、安全、合成速度快、可以短期内大量合成等等,都会让它成为未来疫苗的优先考虑手段,甚至是最佳选择。可以想象未来几年,将会出现各种mRNA疫苗的开发热潮。一旦此类技术成熟,以后如果再出现这样的全球大流行的传染疾病,科学家们将会有例可循,会更加从容地面对,在短期内开发出拯救人类的疫苗来。
?
FDA这个紧急授权,是基于周四专家委员会对于临床试验数据做出的分析结论而下的。辉瑞公司提交的三期有效性临床试验显示有效率达95%,而且没有发现明显的副作用,只有2%-3.7%的受试者出现轻微的疲劳和头疼,显示安全有效。观察到的副作用极少也轻微。大多数表现为轻度的疲劳,低热,注射部位疼痛。严重的副作用很罕见。超过4.4万的临床试验中,只观察到4例参试者出现贝尔面瘫,但都是暂时性的。疫苗注射组观察到64例参试者出现淋巴结肿胀,安慰剂组有6例。实验结束时,这些副作用均得到缓解恢复。

Moderna疫苗实验室中的科学家 |?https://www.geekwire.com
正在FDA审批中的另一家美国的生物公司Moderna开发的mRNA疫苗,其临床三期试验的数据分析也显示有效率达94.1%。对于重度症状患者的有效性达到了100%。有了辉瑞的前例,Moderna的疫苗批准应该也快了。
这是奇迹一样的速度。要知道,普通一个疫苗从开发到上市,平均要5到10年的时间,甚至更长。
时不我待
按照FDA的标准,一个疫苗需要在三期试验中至少达到有效性高于安慰剂组50%以上才能称为清晰且可信(clear and compelling)的有效性。而且统计分析要显示这个有效性有可靠的数据支持,不是因为碰巧的运气所致。当然,有效性越高越好。但FDA此前的记录显示,有效性高过50%就有望获批。
通常情况下,厂家做完临床试验需要收集整理数据,然后向FDA递交生物制品许可申请(Biologics License Application),申请批准市场化临床使用。这些多达几千页的实验数据整理成文通常要费时几个月。FDA拿到申请后,需要组织专家组对申请数据进行详细的分析讨论,最终做出决定是否批准商品化。这个分析讨论到最终做出决定通常也需要几个月的时间。常常还需要组织公众专家讨论会,以咨询行内专家的意见,所以费时费力。因为FDA力求自己做出的决定是科学可靠的,以保持公众对FDA的信任。

亚利桑那州一家医院中,一位新冠病人和医护人员的合影 | www.ft.com
但今年,情况特殊。尤其是正在全球肆虐的新冠肺炎病毒绝非通常的病毒。每天成百上千的人死于该病毒,全球许多国家也因此中断了经济活动,受影响的人数以亿计。所以新的疫苗是全球70亿人翘首以盼的。每多拖一天,就多付出一天的代价。特殊时期,一切都得特殊对待。
所以,FDA提醒厂家可以申请紧急使用授权(Emergency Use Authorization,EUA)。这是美国国会在911恐怖袭击之后,为了应对突发的公众健康紧急状况,给予FDA在特殊时期的特权,可以在正式流程批准之前,紧急批准一些可能拯救大量生命的治疗方案。按照联邦法,要达到EUA的标准,新的治疗方案或者药物要符合这样两条标准:
该药物对于避免和预防严重的威胁生命的疾病有可能的显效。
已知的治疗效果和潜在的公众获益,要大于已知或者可能的毒副作用。
?
当然,法条是死的,每一个提请EUA的药物或者疫苗都需要具体对待。如何定义利大于弊,也是因事而异。比如,一个终末期的病人,其他一切可行的治疗均告无效,这时候如果一个新的治疗方案即便效果不太确定,或者已知的副作用虽然确实存在但在可接受范围内,那这样一个新药就可以算是利大于弊。

加州街头,因恐惧新冠病毒而纷纷戴上口罩的市民 | www.nbcnews.com
疫苗又不一样。因为疫苗一旦批准,将会给成千上万甚至百万千万的健康人接种,所以FDA对于疫苗的EUA非常谨慎。他们强调,只有该疫苗在大人群的严谨的对照双盲三期试验数据显示确实有效, 同时疫情失控难以控制的情况下,FDA才会考虑批准EUA。否则,就得按照正常流程走BLA申请。
这次的新冠疫情显然符合EUA的情况。所以辉瑞公司于11月20日,Moderna于11月30日,分别向FDA递交了EUA申请。21天后的今天FDA就做出了批准,推荐就是EUA授权。
根据FDA的推荐意见公告,辉瑞公司提交给FDA的最终有效数据有37586人,年龄为16岁以上。没有发现任何明显或者严重的副作用。接种组抗病毒有效率达95%。FDA专家组的结论是,该疫苗的益处远远大于可以预期的坏处,利大于弊,有信心批准使用将有利公众健康,因此授权使用。16岁以下的未成年人,因为目前试验数据不够做出评判,暂缓推荐。
据美国疾控中心CDC的公告,下周即将开始的全面接种将优先考虑高风险的人群,包括医务人员,和居住在养老院的老人,接下来会是有慢性病等风险的老人,最后才会是普通人。
具体疫苗接种将会是这样的。运送到各地的疫苗被冷冻保存在玻璃的小瓶子中,使用时用 1.8 毫升的 0.9%的生理盐水稀释。稀释后将可用于5人注射使用。每人注射 0.3 毫升的稀释液。每剂含有30微克的mRNA疫苗,以及极少量的其他保存疫苗活性的成分。每次每人注射0.3毫升疫苗稀释液,隔开3周接受第二剂注射,就算完成接种。
目前为止,美国政府购买了辉瑞疫苗一亿剂,够5千万人接种。最快下周一就有医护人员接受注射接种。从辉瑞总部所在的密西根州厂房外,UPS和FEDEX的货运车已经准备好等了好几天了,就等FDA和CDC的批准下达,立刻开始送往机场,向全美各地运输。
12月17日,FDA还会有针对Moderna疫苗的最终意见。如无意外,也应该是会很快获得批准。这个疫苗比起辉瑞疫苗保存条件要求低,普及会更加容易。
人类抗击新冠病毒终于迎来了里程碑的一刻,胜利的曙光终于在地平线上出现了。
作者:勿怪幸
编辑:朱步冲

新冠疫苗的开发进展迅速,果壳的数据可视化团队DATAMUSE开发了一个新冠疫苗追踪器,展示了全球新冠疫苗的开发阶段、类型以及相关信息。你可以扫描下方二维码或点击下方“阅读原文”访问。我们会和你一起持续关注每一支新冠疫苗的最新进展,直到世界恢复正常的那一天。


本文来自果壳,未经授权不得转载.
如有需要请联系sns@guokr.com

关注公众号:拾黑(shiheibook)了解更多
[广告]赞助链接:
四季很好,只要有你,文娱排行榜:https://www.yaopaiming.com/
让资讯触达的更精准有趣:https://www.0xu.cn/






 果壳
果壳
 关注网络尖刀微信公众号
关注网络尖刀微信公众号随时掌握互联网精彩






- 1 习近平将发表二〇二六年新年贺词 7904141
- 2 2026年国补政策来了 7808738
- 3 东部战区:开火!开火!全部命中! 7712893
- 4 2026年这些民生政策将惠及百姓 7616985
- 5 小学食堂米线过期2.5小时被罚5万 7519709
- 6 解放军喊话驱离台军 原声曝光 7428214
- 7 为博流量直播踩烈士陵墓?绝不姑息 7327605
- 8 每月最高800元!多地发放养老消费券 7238391
- 9 数字人民币升级 1月1日起将计付利息 7141831
- 10 2026年1月1日起 一批新规将施行 7040675